Identificata all’ospedale pediatrico della Santa Sede la mutazione responsabile di una patologia con soli 5 casi al mondo. La guarigione grazie al trapianto di midollo da genitore. Il caso pubblicato sul Journal of Experimental Medicine
Roma, 28 febbraio 2020 – La piccola Diana è nata con una malattia gravissima e sconosciuta, che la mette in continuo pericolo di vita. Un percorso lungo e doloroso per lei e per la sua famiglia, presa in carico dall’Ospedale Pediatrico Bambino Gesù di Roma. Dopo anni di studi e di tentativi, i ricercatori riescono a identificare la mutazione del gene responsabile della sua patologia ultra-rara, di cui ad oggi si conoscono solo altri 4 casi al mondo, quasi tutti purtroppo con esito drammatico.
Ma i medici non si arrendono e grazie a un trattamento farmacologicosperimentale e a un trapianto di midollo, riescono a salvarle la vita. Oggi Diana sta bene ed è finalmente guarita. La sua storia clinica e i risultati della ricerca sono stati pubblicati sul Journal of Experimental Medicine. Alla vigilia della giornata mondiale delle malattie rare, il Bambino Gesù sottolinea l’importanza della ricerca scientifica e dell’approccio multidisciplinare per riuscire a dare un nome alle malattie senza diagnosi. Il primo passo verso una possibile terapia.
La scoperta del gene malattia
Diana viene trasferita al Bambino Gesù a due settimane dalla nascita. Le sue condizioni appaiono da subito gravi e la malattia inspiegabile: la pelle piena di macchie, febbre alta continua, gravi carenze di cellule nel sangue (globuli rossi, globuli bianchi, piastrine). Seguita dai medici di onco-ematologia e reumatologia, la bambina passa i primi sette mesi di vita in isolamento in Ospedale, nel tentativo di identificare la sua malattia o almeno tenerla sotto controllo. Il primo successo arriva con un farmaco biologico (anakinra), che riesce finalmente a contenere gli eccessi infiammatori provocati dalla patologia ancora sconosciuta. La bambina migliora e per la prima volta può tornare a casa.
Dopo pochi mesi, purtroppo, la situazione peggiora improvvisamente. Diana presenta sintomi nuovi tra cui nuove riaccensioni auto-infiammatorie, emorragie intestinali e crisi convulsive. I medici sono costretti a cambiare terapia, ma la storia prosegue all’insegna della preoccupazione e dell’incertezza, tra ricadute e ricoveri continui. Non è però tempo perso: la bambina nel frattempo viene inserita nel programma di ricerca dedicato alle malattie rare e senza diagnosi basato sull’uso delle nuove tecnologie genomiche, finanziato dalla campagna Vite Coraggiose della Fondazione Bambino Gesù onlus.
Grazie alle piattaforme di sequenziamento di ultima generazione, i ricercatori arrivano a identificare una mutazione potenzialmente implicata nella malattia, su un gene chiamato CDC42. Un gene di cui erano state precedentemente individuate dagli stessi ricercatori dell’Ospedale altre mutazioni associate a diverse malattie del neurosviluppo.
Inizia così la ricerca incrociata sui database mondiali di malattie rare. È il 2016 quando si scopre un altro paziente con lo stesso quadro clinico e la stessa mutazione negli Stati Uniti, purtroppo già deceduto. Poco dopo vengono scoperti altri due pazienti con la stessa mutazione: uno al Karolinska Institutet di Stoccolma e uno ancora al Bambino Gesù. Compreso quello di Diana, sono 4 i casi al mondo noti in questo momento (un altro paziente, il quinto, verrà identificato da un team di ricercatori statunitensi).
“Identificare una possibile mutazione come causa di malattia rappresenta però solo il primo passo – spiega uno dei coordinatori dello studio, dott. Marco Tartaglia, responsabile del laboratorio di Genetica Molecolare e Genomica Funzionale – Per dimostrarne il ruolo causativo è infatti necessario attivare una serie di studi di validazione funzionale che consentano di capire in che modo la mutazione identificata possa alterare la funzione della proteina e conseguentemente i processi cellulari e fisiologici implicati nella malattia”.
L’identificazione di più pazienti con questa mutazione e con quadri clinici sovrapponibili e la dimostrazione dello specifico impatto funzionale della variante arriva dopo due anni di ricerche, mentre Diana continua la sua lotta per la vita. Gli studi permettono di confermare che la mutazione del gene CDC42 è effettivamente la causa della sua malattia, e la malattia può finalmente avere un nome: sindrome NOCARH (Neonatal-Onset Cytopenia with dyshematopoiesis, Autoinflammation, Rash and Hemophagocytosis).
Ora è possibile concentrarsi sulla ricerca di una terapia: differentemente dalle altre malattie causate da mutazioni in CDC42, la condizione di Diana è essenzialmente limitata alle cellule del sangue. Sulla base di questa considerazione e dei dati prodotti dalla ricerca, si ritiene che il trapianto di midollo, quindi la sostituzione delle sole cellule del sangue, possa sconfiggere la malattia.
Un percorso a ostacoli verso il trapianto
La patologia di Diana, che comincia a essere via via più comprensibile, continua a manifestare delle fasi infiammatorie acute molto gravi, che possono portare anche alla morte, come già accaduto agli altri pazienti.
Nella primavera del 2017 la bambina presenta una di queste ricadute infiammatorie con un infarto intestinale. Nonostante la sua condizione, si decide di sottoporla a intervento chirurgico per scongiurarne la morte. Dopo 4 ore, la bimba esce dalla sala operatoria e torna in rianimazione. Ce l’ha fatta, ma le sue condizioni risultano più critiche che mai: ha sviluppato infatti una iper-infiammazione che fa temere ai medici il peggio.
I clinici decidono di usare, in via compassionevole, un altro farmaco sperimentale, l’emapalumab, usato fino ad allora solo su 15 bambini prima di Diana (nessuno dei quali con la sua malattia). Si tratta di un anticorpo monoclonale che serve a controllare l’esasperata risposta infiammatoria nei pazienti con HLH (Linfoistiocitosi Emofagocitica primaria).
L’Ospedale Bambino Gesù coordina le sperimentazioni nel mondo di questo farmaco, che poi sarà usato con successo per Alex, il bambino trasferito a Roma da Great Ormond Street Hospital di Londra nel novembre 2018 e accompagnato al trapianto di cellule staminali emopoietiche dal prof. Franco Locatelli, direttore del dipartimento di Onco-ematologia e terapia cellulare e genica dell’Ospedale della Santa Sede.
Il farmaco funziona anche con Diana, persino oltre le aspettative: la fase acuta passa in pochi giorni e la bambina sta subito meglio. “Diana aveva già dovuto superare momenti di grande difficoltà a rischio di vita – ricorda il dottor Fabrizio De Benedetti, responsabile di Reumatologia del Bambino Gesù – I risultati delle lunghe ricerche in laboratorio ci hanno permesso di applicare questa terapia in modo personalizzato, mirando al meccanismo alla base della sua malattia”.
È possibile finalmente procedere con la fase finale della terapia: il trapianto di midollo. Diana non ha però un donatore compatibile e si deve quindi ricorrere a uno dei due genitori (trapianto aploidentico), in questo caso il papà: si tratta di una procedura complessa basata sulla manipolazione delle cellule staminali emopoietiche prelevate dal donatore, per privarle selettivamente di tutti gli elementi che potrebbero aggredire l’organismo del ricevente.
“Un trapianto di cellule staminali emopoietiche da donatore aploidentico su una paziente con una patologia fino ad oggi sconosciuta presentava diverse incognite, ad esempio l’eventualità di un non attecchimento delle cellule infuse – spiega il professor Franco Locatelli – Il rischio di insuccesso era altissimo, ma si trattava anche dell’unica opzione per sconfiggere la malattia con cui Diana lottava dalla nascita”.
Un intervento unico al mondo per una bambina unica. Il trapianto è stato un successo e oggi Diana è guarita, non presenta più segni della malattia. Una malattia di cui non si sapeva il nome e di cui la bambina conserverà, forse, soltanto il ricordo.
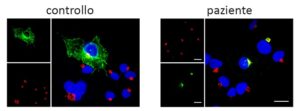
Foto gene CDC42: Differentemente da quanto si osserva nelle cellule di soggetti sani (pannello a sinistra), la mutazione che causa la sindrome NOCARH blocca la proteina CDC42 (colorata in verde) in uno specifico comparto intracellulare (colorato in rosso), come mostrato nel pannello di destra
 Salva come PDF
Salva come PDF